Homocisteina
cap22
http://www.medicalnewstoday.com/articles/318526.php?utm_source=newsletter&utm_medium=email&utm_campaign=daily-hcp
Genetica
A forma mais comum é a mutaçao do gene responsavel pela sintese ds cistationina sintetase que catalisa a transformaçao da homocisteina em cistationina


Genetica
A forma mais comum é a mutaçao do gene responsavel pela sintese ds cistationina sintetase que catalisa a transformaçao da homocisteina em cistationina
Acumula-se homocisteina e por remetilação desta metionina
A homocisteina pode reagir com os grupos lisil do colagenio
Hereditariedade
É uma doença autosomica recessiva
Hereditariedade
É uma doença autosomica recessiva
http://www.newbornscreening.info/Parents/aminoaciddisorders/CBS.htm
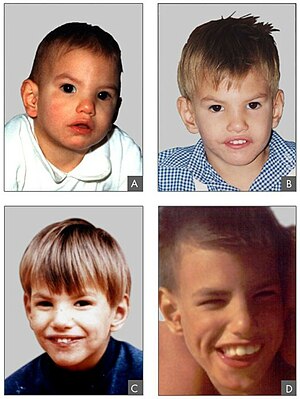
Sintomas da formas mais comum
- Miopia
- Descolamento do cristalino
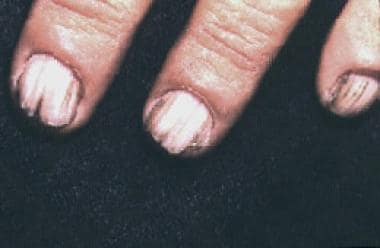
- Coagulaçao do sangue anormal
- Osteoporose
- Atrasos no crescimento
- Dificuldades na aprendizagem
Sintomas das formas mais raras
- Atrases no crescimento
- Mau desenvolvimento intelectual
- Convulsoes
- Dificuldade nos movimentos
- Anemia megaloblastica
Frequencia
Na forma mais comum varia entre 1/200.000 e 1/335.000
Tratamento
Tentou-se como terapêutica a restrição de metionina e a alimentação com betaina ou colina
Nalguns casos obtiveram-se bom resultados com vitamina B6.Os doentes podem dividir-se em dpois grupos: B6 sensiveie e B6 resistentes
Actualmente há dados consistentes sugerindo a administração de folato como via de tratamento
A betaina baixa a cisationina no sangue
A betaina baixa a cisationina no sangue
Defeitos na formação de metilcobalamina


Cinco defeitos enzimáticos diferentes podem interferir na formação de metilcobalamina
Os doentes têm vómitos, letargia, hipotonia e atrasos no desenvolvimento
O laboratório revela uma anemia megaloblastica
Tratam-se com a administração de vitamina B12
Bibliografia
Deficiência em metileno- tetrahidrofolato redutase
Causa
Inibiçao da transformaçao do 5-10 metilenç tetrahidrofolico bem 5-metilo tetrahidrofolico, cosubstrato da remerilaçao da homocisteinsna em metionina
Causa
Inibiçao da transformaçao do 5-10 metilenç tetrahidrofolico bem 5-metilo tetrahidrofolico, cosubstrato da remerilaçao da homocisteinsna em metionina
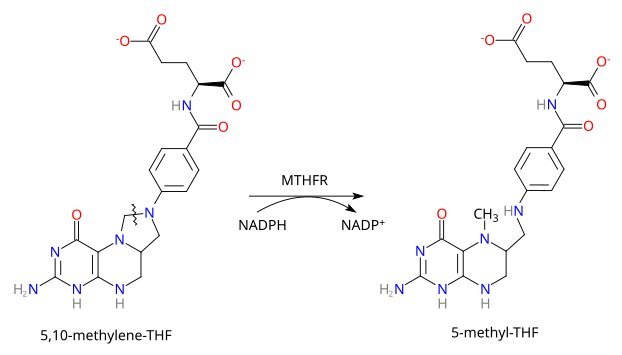
http://en.wikipedia.org/wiki/Methylenetetrahydrofolate_reductase
Genetica
Deve-se ao defeito de um gene colocado no cromosoma 1
Sintomas
Na deficiência total há apneia neonatal e convulsões que podem levar rapidamente à morte
Na deficiência parcial observa-se um quadro crónico com atraso mental, convulsões,microcefalia e espasticidade
Tretamento
Tretamento
Trata-se com a administração precoce de betaina
Os sintomas psicoticod tratam-se com a administraçao de piridoxina e acido folico
Os sintomas psicoticod tratam-se com a administraçao de piridoxina e acido folico
Bibliografia
cap22
Homocistinemia e ateroesclerose
A homocistenemia é um factor de risco da ateroesclerose
A homocisteina em excesso forma a sua tiolactona que reage com os aminoacidos livres das LDL provocando a sua agregação e endocitose pelos macrofagos
Provoca ainda oxidação dos lipidos e agregação das plaquetas
Hipermetioninemia
Bibliografia
Secundaria
Observa-se em doenças hepáticas na tirosinemia tipo I e na homocistinuria clássica
Pode observar-se na imaturidade transitória da metionina adenosiltransferase em recemnascidos alimentados com dietas ricas em proteínas
Primaria
Deficiencia da adenosina metiltransferase hepática
Pode haver desmielinização
Sintomas
Na maior parte dos casos não há sintomas.
Nalguns pode-se observar:
Hereditariedade
É uma doença autosomica recessiva, embota em certos casos possa ser dominante
Sintomas
Na maior parte dos casos não há sintomas.
Nalguns pode-se observar:
- Diminuiçao das capacidades intelectuai
- Problemas neurologicos
- Fraqueza muscular
- Caracteristicas faciais proprias
- Problemas hepaticos
- Respiraçao, suor e urina com cheiro proprio
Hereditariedade
É uma doença autosomica recessiva, embota em certos casos possa ser dominante
Cistinuria
Genetica
Doença autosomica recessiva da reabsorção tubular dos aminoacidos dibasicos cistina, ornitina, lisina e arginina
Hereditariedade
Doença autosomica recessiva

Hereditariedade
Doença autosomica recessiva
Prevalencia
1/7.000
Sintomas
A cistina é relativamente insolúvel, podendo nos homozigotos, precipitar e formar cálculos renais
Na maior parte dos casos nao há sintomas

http://www.medicinenet.com/cystinuria/article.htm
Tratamento
Na maior parte dos casos nao há sintomas
http://www.medicinenet.com/cystinuria/article.htm
Tratamento
- Alcalinizaçao da urina
- Reduçao do consumo de sal e proteinss
- Quelaçao
- Cirurgisa
Bibliografia
cap22
Cistinose
Patologis
É ums doença autosomica recessiva devido à não produçao de cistinosina, transportador que leva a cistina dos lisosomas pata os tecidos, levando assim à acumulaçao de cistinaem todos os tecidos.
Há tres tipos diferentes de cistinose - ocular, intermediaria e renal
Cistinose rena
Como consequencia do deposito no rim de ums grande quantidade de cistin há uma grande perda de sais minerais o que provoca um sindroma conhecido por sindroma de Fanconi - raquitismo hipofosfatemico, poliuria,sede,desidrataçao e acidose
É ums doença autosomica recessiva devido à não produçao de cistinosina, transportador que leva a cistina dos lisosomas pata os tecidos, levando assim à acumulaçao de cistinaem todos os tecidos.
Há tres tipos diferentes de cistinose - ocular, intermediaria e renal
Cistinose rena
Como consequencia do deposito no rim de ums grande quantidade de cistin há uma grande perda de sais minerais o que provoca um sindroma conhecido por sindroma de Fanconi - raquitismo hipofosfatemico, poliuria,sede,desidrataçao e acidose
A partir dos 2 anos, os depositos de cistina nos olhos provocam fotofobia
Em doentes não tratados pode surgir deterioraçao muscular, cegueira, impossibilidade de engolir, ausencia de sudaçao, pigmentaçao da pele, diabetres, problemas na tiroide e sistema nervoso.
Cistinose intermedia
Os sintomas sao os mesmo da nefropatica mss eurgem numa idade mais tardia - 12-15 anos
Tratamento
Em doentes não tratados pode surgir deterioraçao muscular, cegueira, impossibilidade de engolir, ausencia de sudaçao, pigmentaçao da pele, diabetres, problemas na tiroide e sistema nervoso.
Cistinose intermedia
Os sintomas sao os mesmo da nefropatica mss eurgem numa idade mais tardia - 12-15 anos
Tratamento
- Cisteamina
- Citrato de sodio para a acidose
- Transplantaçao do rim em casos graves
cap22